![]()
|
Genética
de la Enfermedad de Alzheimer: Situación en el año 2000
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Dr. Jordi Perez-Tur Doctor en Biología Institut de Biomedicina de València-CSIC Dirección para correspondencia: Dr. Jordi Pérez i Tur. Unitat de Genètica Molecular. Institut de Biomedicina de València. C/ Jaume Roig, 11. E-46010 València. Teléf. 96 339 1760 ext 115. Fax 96 369 0800Correo-e jpereztur@ibv.csic.es |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
I. Objetivo. En los últimos 10 años, el estudio de la enfermedad de Alzheimer (EA) ha revolucionado nuestra concepción de la misma, llegando ahora a tener, a pocos años vista y por primera vez, la esperanza de tratamientos efectivos. Una parte de estos conocimientos ha venido de la mano de la aplicación de las técnicas de genética molecular al estudio de la enfermedad. La identificación de genes causantes de la EA ha permitido establecer hipótesis, discutibles como toda hipótesis, sobre las causas y el curso de la patología. En esta ponencia pretendo hacer una revisión sobre los avances más recientes que se están produciendo en el estudio de la enfermedad, en especial sobre la identificación de dos nuevos loci en los que se espera encontrar nuevos genes relacionados con la enfermedad así como sobre el análisis de patrones genéticos que confieran un mayor riesgo por la enfermedad. De manera general, las abreviaturas correspondientes a genes y proteínas seguirán la nomenclatura internacional, los genes se representan en mayúsculas y cursiva mientras las proteínas se representan únicamente en mayúsculas. II. El
siglo XX. Los estudios patológicos sirvieron para caracterizar la
enfermedad de Alzheimer como una enfermedad debida a la muerte neuronal
en áreas límbicas del cerebro y acompañada de la aparición de depósitos
intra y extracelulares de proteínas. Un análisis detallado de estos depósitos
demostró la implicación mayoritaria de dos proteínas que se convirtieron,
desde entonces, en el centro de la investigación sobre la EA. De una
lado, los depósitos intracelulares tenían como componente mayoritario
acúmulos de proteína TAU con un patrón de fosforilación diferente al normal[Kosik,
1986 #3369]. Con posterioridad, se ha observado que depósitos similares
aparecen en un amplio abanico de trastornos degenerativos del sistema
nervioso, excelentemente revisados por Buée y Delacourte[Buee, 2000 #3370].
De otro lado, los depósitos extracelulares están constituidos por un gran
número de proteínas de entre las que destaca, por ser la más abundante,
un péptido de entre 39 y 42 aminoácidos, el péptido amiloide ó Ab. Como
su nombre indica, el péptido Ab tiene una fuerte tendencia a polimerizar
formando fibras insolubles (amiloide) que son las que se depositan constituyendo
una estructura con una fuerte tendencia a atrapar otras moléculas que
circulen por el espacio extracelular.El péptido Ab aparece como el resultado
del procesamiento celular de un precursor, la proteína precursora del
amiloide (APP) que puede sufrir diversos cortes proteolíticos (figura
1) por unas enzimas recientemente identificadas (ver más adelante). En
determinadas circunstancias se favorece la vía que conduce a la formación
del péptido Ab (vía amiloidogénica), mientras que normalmente, el procesamiento
de la APP sigue la llamada vía no amiloidogénica que evita la aparición
de este péptido. No se conoce la función de la APP, ni del péptido amiloide,
si bien las neuronas lo producen normalmente aunque en muy baja concentración
. Uno de los puntos clave de este proceso ha sido la identificación de
las enzimas que procesan a la APP para producir el péptido Ab puesto que
se consideran como las dianas terapéuticas más susceptibles de ser efectivas.
Si bien la a-secretasa es todavía desconocida, en el último año, varios
grupos han logrado identificar las enzimas responsables de las actividades
b- y g-secretasa[Sinha, 1999 #3373; Yan, 1999 #3374; Marshall, 1999 #3375;
Hussain, 1999 #3376; Vassar, 1999 #3372; Li, 2000 #3181], con lo que se
abren nuevas vías de investigación terapéutica.Existe un nivel adicional
de complejidad en este proceso que es debido a que la síntesis del péptido
Ab no produce una población de péptidos homogéneos. La mayor parte del
producto de procesamiento de la APP por la vía amiloidogénica lo constituye
un péptido de 39-40 aminoácidos de longitud, mientras que una fracción
minoritaria la forma un péptido de 42-43 aminoácidos. Esta mínima diferencia
en talla tiene un efecto importante en cuanto a la formación de las fibrillas
del amiloide. La forma más larga presenta una mayor insolubilidad y,
por consiguiente, tiende a formar fibrillas de manera más rápida que la
forma corta.El descubrimiento de que las placas seniles estaban constituidas
mayoritariamente por el péptido amiloide puso en el centro de mira al
gen que lo codifica como factor responsable de la EA. De hecho, estudios
genéticos realizados al final de los años 80, demostraron que un grupo
de familias con la forma de inicio precoz (anterior a 65 años) de la enfermedad
segregaba de manera autosomal dominante y con una penetrancia alta mostraban
ligamiento a un locus en el cromosoma 21 en el que se encontraba,
entre otros genes, el gen de la APP[St George-Hyslop, 1987 #2049]. Por
otra parte, una prueba indirecta de la influencia que este locus
debía tener vino del descubrimiento de que los pacientes con trisomía
21 también desarrollaban síntomas clínicos y patológicos de EA si, en
la duplicación del cromosoma 21 causante de la trisomía, se incluía el
locus en el que se encuentra APP[Prasher, 1998 #3397].
El análisis posterior de este locus demostró que aquellas familias en
las que la enfermedad se encontraba ligada al cromosoma 21 presentaban
mutaciones en el gen de la APP[Goate, 1991 #2052; Chartier-Harlin, 1991
#2053]. Desde 1991, año en el que se describieron las primeras mutaciones,
se han encontrado pocas mutaciones en este gen, se le considera responsable
del 5-10% de las formas familiares autosómica dominante de inicio precoz
(una lista actualizada de mutaciones en este gen, con las referencias
bibliográficas correspondientes, se puede encontrar en la siguiente página
de Internet: http://www.alzforum.org/members/resources/app_mutations/).Poco
tiempo después del descubrimiento de la existencia de un locus
ligado a la EA en el cromosoma 21, comenzó a observarse que una parte
importante de las familias en las que la EA segregaba de manera autosómica
dominante y con una edad de inicio precoz no solamente no presentaban
ligamiento con ese locus sino que, incluso, éste quedaba excluido[Schellenberg,
1988 #2594]. A posteriori se ha observado que las familias no
ligadas al cromosoma 21 eran justamente aquellas en las que la enfermedad
tenía tendencia a aparecer a edades más tempranas (la edad media de aparición
de muchas de esas familias se encontraba en el inicio de la 4ª década
de vida), y con una penetrancia más elevada, es decir, aquellas formas
de la enfermedad que podían considerarse más agresivas no presentaban
ligamiento con el locus en el que se encuentra el elemento central
de la patología.La controversia que siguió a estos descubrimientos se
saldó con la identificación de un segundo locus para la enfermedad en
el cromosoma 14, concretamente en la región 14q24.3[Schellenberg, 1992
#2055], trabajo inmediatamente confirmado por otros grupos [Mullan, 1992
#2056; St George-Hyslop, 1992 #2057]. Al igual que sucedió con la localización
del primer locus para la EA en el cromosoma 21, el primer trabajo
describiendo un locus en el cromosoma 14 ya demostró la existencia de
un tercer locus causante de una forma de inicio precoz y herencia autosómica
dominante en una familias que, por su origen histórico-geográfico, se
conocen como familias Germano-Volga. Este es un grupo de familias
de origen germano que emigraron al valle del Volga, en Rusia, antes de
que una parte ellas emigrasen, a su vez, a los EE.UU. donde fueron clínica
y molecularmente estudiadas por presentar una forma de EA de herencia
mendeliana e inicio precoz, similar en cuanto a la edad de inicio a las
formas ligadas al cromosoma 21 si bien se excluyó ligamiento a la APP
y al nuevo locus en el cromosoma 14[Schellenberg, 1992 #2055].En 1995
se identificó el gen responsable de las formas ligadas al cromosoma 14
[Sherrington, 1995 #2058] que se denominó presenilina 1 (PSEN1)
al ser el causante de las formas preseniles. El ordinal se utilizó al
comprobarse que este gen tenía un homólogo en el cromosoma 1, PSEN2,
que también estaba genéticamente ligado a la EA, precisamente en las formas
de origen germano-Volga[Levy-Lahad, 1995 #2738]Estos dos genes son altamente
homólogos en su secuencia y, por lo tanto, en su estructura proteica primaria.
A pesar de su gran similitud, su importancia en relación con la EA, medida
como número de mutaciones en diferentes formas familiares de la enfermedad,
es extremadamente distinta. Mientras el número de familias que presentan
mutaciones asociadas a la PSEN1 es muy elevado (contabilizándose
del orden de 60 mutaciones distintas), en el caso de la PSEN2 el
número de mutaciones descritas en la bibliografía es más reducido, situándose
a un nivel equivalente al de las mutaciones en el gen de la APP[Cruts,
1998 #2747] (una lista actualizada de mutaciones en estos genes puede
encontrarse en la siguiente dirección en Internet: http://www.alzforum.org/members/resources/pres_mutations/index.html
con enlaces a las correspondientes referencias bibliográficas).Al contrario
de lo que sucede en el caso de la APP, donde las mutaciones se
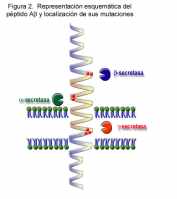
localizan alrededor de los puntos de corte de la Ab (figura 2), las mutaciones
no parecen localizarse en ninguna área definida de las PSEN (figura
3), además las mutaciones afectan a residuos altamente conservados entre
las presenilinas de varias especies, indicando su importancia desde el
punto de vista evolutivo[Hutton, 1997 #3371].Recientemente, se ha identificado
la relación entre las PSEN y la EA[Li, 2000 #3181]. Los elegantes
experimentos de fotomarcaje de Li y sus colaboradores demostraron claramente
el papel de PSEN1 y PSEN2 como poseedoras de la actividad
enzimática que provoca el corte III. Del siglo XX al siglo XXI. El análisis genético realizado en los años 80 y 90 ha permitido identificar 3 genes causantes de la enfermedad, conocer a través de ellos la importancia de la producción del péptido Ab y determinar algunas vías interesantes en cuanto a tratamiento. Sin embargo, la mayor parte de los individuos afectados por la EA no pertenecen a grupos familiares de inicio precoz si no que constituyen el grupo de inicio posterior a los 65 años, inicio tardío. En este caso, la influencia de la historia familiar resulta difícil de estimar puesto que resulta complicado el determinar con la precisión necesaria el estado cognoscitivo de los familiares directos del paciente así como por el hecho de que, al tratarse de enfermedades que se manifiestan de manera tardía en la vida, frecuentemente los familiares de estos pacientes pueden haber fallecido por otras causas no relacionadas con ningún proceso neurodegenerativo. A pesar de esto, los estudios epidemiológicos realizados sugieren que la forma senil de la EA tiene un componente genético importante.En ocasiones, el componente genético puede seguir un patrón de herencia mendeliana pero frecuentemente el componente hereditario no sigue estas leyes de herencia debido a que se trata de una enfermedad multifactorial. Con este término se engloban aquellas patologías cuyo origen depende de la existencia simultánea de varios factores, genéticos y/o ambientales. En este sentido, la EA de inicio tardío puede aparecer como el resultado de la presencia de variabilidad genética en diversos genes de manera que cada uno de los alelos implicados, de manera aislada, no tengan ningun efecto en cuanto a la presencia de la enfermedad y, únicamente en combinación con otros alelos en genes distintos es cuando las vías sobre las que actúan llegan a verse comprometidas originándose la EA.En 1991, Pericak-Vance y sus colegas de la Universidad de Duke determinaron la existencia de un locus en el cromosoma 19 relacionado con formas familiares de EA de inicio tardío[Pericak-Vance, 1991 #2087]. Más adelante se identificó al gen responsable del ligamiento y se demostró que se trataba de un gen conocido con anterioridad por su relación con el metabolismo del colesterol, la apolipoproteína E (APOE) [Strittmatter, 1993 #1760]. Este gen presenta 3 isoformas en la población general que se corresponden con 3 alelos a nivel del gen. La diferencia entre las isoformas es mínima, de 1 ó 2 aminoácidos, sin embargo este ligero cambio tiene un efecto a nivel de la función de la proteína que se refleja en la existencia de asociaciones genéticas entre varias de las isoformas y diversas patologías. En el caso de la EA, se ha observado que la posesión de, al menos, una copia del alelo e4 produce un aumento del riesgo frente a la enfermedad que oscila entre 4 y 20 veces en función de la población en la que se estudie su efecto. Además se vió que el riesgo conferido por la posesión de este alelo era dependiente de la dosis génica[Corder, 1993 #2343], tanto en cuanto al riesgo por la EA asociado a su posesión como a su efecto sobre la edad de inicio de la misma[Corder, 1993 #2343]. Aquellos individuos homozigotos para el alelo e4 presentan una edad de inicio más temprana y un riesgo mayor. Aunque la relación fisiológica entre la APOE y la EA aún no se conoce con precisión, parece que ésta se da a través de la vía de formación de amiloide insoluble puesto que se ha observado una relación entre la dosis génica en este locus y la cantidad de depósitos de amiloide observados en el parénquima cerebral de los afectados, aquellos enfermos con una copia del alelo e4 presentan un mayor número de depósitos que aquellos enfermos sin ninguna copia de este alelo, mientras que los individuos e4 homozigotos son los que presentan una mayor densidad de placas seniles[Schmechel, 1993 #1614].Sin embargo, ni la posesión de una copia del alelo e4 de la APOE determina la aparición de la enfermedad, ni la no presencia de dicha variante garantiza su ausencia de forma absoluta. En este caso estamos hablando de factores de riesgo genéticos que deben manifestarse en función de la coincidencia con otros factores para dar lugar a la enfermedad. En este sentido cabe destacar el trabajo realizado en los 2 últimos años referentes a la caracterización de la relación entre el gen de la APOE y la EA mediante el estudio de variabilidad genética localizada en áreas importantes desde el punto de la regulación de la expresión de este gen [Lambert, 1997 #2090; Artiga, 1997 #2092].Diferentes estimaciones sugieren que la APOE puede tener un efecto importante en, aproximadamente, el 50% de la población de pacientes. Otros estudios indican que el efecto de la APOE pueda estar más relacionado con el desarrollo del proceso patológico más que con su aparición, incidiendo especialmente en la edad de inicio de la enfermedad en aquellos individuos con un riesgo a padecerla en función de otros factores genéticos o ambientales[Meyer, 1998 #2065].En cualquier caso es evidente que la susceptibilidad genética por la EA de inicio tardío es más compleja y que hay otros factores aún por identificar. En los últimos tiempos, se ha producido un aumento espectacular del número de estudios de asociación genética realizados dirigidos a la identificación de nuevos factores de riesgo genéticos por la enfermedad. En estos estudios, y partiendo de aquello que se conoce, se lanzan hipótesis implicando un determinado gen en la vía patogénica y se ensaya su implicación utilizando diseños de tipo caso-control. La consecuencia de estos trabajos es un flujo continuo de publicaciones en las que se demuestra la existencia de una asociación genética entre el gen estudiado y la enfermedad en una población particular seguido por un número mayor de trabajos en los que o bien se confirman estos resultados o bien se indica la falta de replicación. La consecuencia final de este proceso es asimilable a un estado de confusión permanente donde los distintos grupos interesados en el estudio genético se dedican a la replicación de resultados de otros grupos con un éxito relativo. En la siguiente tabla se muestran unos pocos ejemplos de genes sobre los que se han publicado resultados contradictorios: Tabla 1. Genes estudiados por su hipotética relación con la enfermedad de Alzheimer.
IV. ¿Qué traerá el siglo XXI? Hasta este momento, conocemos la implicación de 4 genes en el desarrollo de la EA aunque no conozcamos con precisión los mecanismos por los que estos genes actúan. Tres de estos genes, APP, PSEN1 y PSEN2, son responsables de una minoría de formas de la enfermedad, las formas de inicio precoz y herencia autosómica dominante. El cuarto gen, APOE, está relacionado con las formas de inicio tardío, tanto las familiares como las formas esporádicas (sin antecedentes familiares). Evidentemente estos genes no explican en su totalidad la influencia genética en la EA, aún existe una parte del riesgo genético del que desconocemos su origen. Un trabajo teórico publicado recientemente sugiere que pueden existir otros 4 loci relacionados con la enfermedad que pueden tener un efecto similar, o mayor, que el de la APOE[Warwick Daw, 2000 #3396]. Sin embargo, hasta el momento, no hemos sido capaces de proporcionar las pruebas de que realmente existan. En los últimos años se ha dedicado un esfuerzo importante a la determinación de los factores genéticos que influencian la aparición de la enfermedad de inicio tardío centrándose, especialmente, en aquellas formas con antecedentes familiares. Estos estudios utilizan una metodología que requiere la colección de muestras de pacientes muy importantes (600-800 individuos afectados) y necesita de un tiempo respetable antes de proporcionar resultados tangibles. De los varios trabajos realizados cabe destacar la identificación de dos nuevos loci en los que se cree pueden existir genes relacionados con la EA.El primer locus se identificó en 1997 en el cromosoma 12[Pericak-Vance, 1997 #2064]. Inmediatamente, otros grupos intentaron confirmarlo[Wu, 1998 #2063; Rogaeva, 1998 #2093] y se observó que, si bien los tres estudios coincidían en la existencia de dicho locus, cada uno lo localizaba en una región diferente del cromosoma. Además, en dos de estas regiones se encontraban genes que ya se habían relacionado genéticamente con la EA: la a2-macroglobulina y la proteína similar al receptor de lipoproteínas de baja densidad si bien con resultados contradictorios (A2M y LRP respectivamente; ver tabla 1). Actualmente se admite la existencia de un gen implicado en la EA en este locus, si bien se desconoce cual de los genes que se localizan en esta área pudiera ser.El segundo locus ha sido descrito aún más recientemente, en el VII congreso internacional de la Enfermedad de Alzheimer y patologías relacionadas celebrado en Wahington D.C. (EE.UU.) en Julio de 2000, y con varios trabajos en fase de revisión. Este locus, en el cromosoma 10, parece suscitar un mayor grado de acuerdo en la comunidad científica pero, desgraciadamente, aún nos encontramos en estadíos muy tempranos del análisis de esta región para poder predecir cuál será el gen implicado en la enfermedad. Los diferentes trabajos mencionados en la tabla 1, muestran la dificultad de encontrar factores de riesgo genéticos clara y reproduciblemente asociados con la enfermedad. Esto es así en parte por que en diferentes poblaciones es probable que actúen diferentes factores de riesgo. Pero también es posible que nos encontremos ante un problema puramente estadístico. Puesto que la enfermedad aparece en función de la presencia, o ausencia, de unos determinados factores de riesgo genéticos, éstos sólo serán detectables en su conjunto en una muestra de tamaño importante de afectados. Por otra parte, los métodos de análisis estadístico que se utilizan corrientemente no permiten, en estos momentos, detectar la influencia de varios genes de manera simultánea, por lo que es necesario el desarrollo de nueva metodología así como su validación antes de ser capaces de establecer estos patrones de susceptibilidad genética. V. Perspectivas.
Como hemos visto, el estudio de las causas genéticas de la EA ha ido evolucionando
a medida que se ha avanzado en su conocimiento. Después de conocer los
genes relacionados con las formas mendelianas, se pasó a la identificación
de factores de riesgo y, ahora, se intenta conocer la existencia de patrones
genéticos que puedan influenciar en la aparición o el desarrollo de
la enfermedad. Con la finalización del proyecto de secuenciación del
genoma humano se abre ahora una doble vía en la investigación. Por una
parte, se debe comenzar la ingente tarea de descifrar a nivel fisiológico
esta enorme fuente de información, qué proteínas se encuentran codificadas
en el genoma y, objetivo final, cuál es su función y en que procesos intervienen.
Por otra, debemos ser capaces de construir el mapa de variabilidad
genética humana que determina tanto el riesgo frente a determinandas
enfermedades como el curso de evolución de las mismas y, finalmente, la
respuesta frente al tratamiento. En este sentido, se habla ya de la medicina
del futuro como de medicina personalizada. Según esta idea, el
ADN de un individuo encierra las claves que permitirán predecir las causas
de la enfermedad e intervenir única y exclusivamente en dichas causas
pero además evitando efectos secundarios adversos puesto que se conocerán
perfectamente las dianas de los fármacos utilizados.Sin embargo, hasta
que esto llegue, se continúa trabajando en los laboratorios en descifrar
las claves genéticas de la EA, para ello se requiere una integración cada
vez mayor entre grupos de investigación clínica y básica así como la colección
de muestras de enfermos de tamaño adecuado para ser capaces de detectar
las sutiles influencias de muchos genes en el desarrollo de la enfermedad.
Iniciativas como las potenciadas desde los Institutos Nacionales de la
Salud de EE.UU. comienzan a rendir sus frutos de manera espectacular,
bien que tarde, se hace necesario aplicar estrategias similares en el
sistema sanitario nacional aprovechando las enormes ventajas que presenta
un sistema de asistencia sanitaria quasi-universal. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
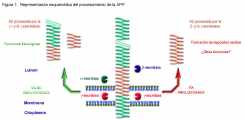 |
Figura 1. Representación esquemática del procesamiento de la APP. La APP, representada aquí en forma de cintas verdes y rojas, se encuentra localizada en la membrana. La formación del péptido Ab (fragmento rojo dentro de la APP) se representa en la parte derecha de la figura y se conoce como vía amiloidogénica, por la acción de la b- y la g- secretasas (representadas en azul y rojo, respectivamente, en la figura), se produce el péptido amiloide que es liberado al medio extracelular bien directamente si el corte se da en la membrana plasmática, bien a través de un proceso exocítico cuando el corte se lleva a cabo en vesículas intracelulares. La vía no amioloidogénica, parte izquierda de la figura, provoca el corte del péptido Ab en dos fragmentos, por la acción de dos enzimas, la a- y la g-secretasa (representadas en verde y rojo en la figura). En este caso, la a-secretasa corta dentro del dominio de la Ab de manera que se produce una proteína que abarca el dominio extracelular de la APP unida al fragmento animo terminal del péptido Ab, la acción posterior de la g-secretasa provoca la aparición del fragmento carboxilo terminal del péptido Ab. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Figura 2. Representación de la estructura del péptido Ab
. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Figura 3. Representación de la estructura de la PSEN1.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
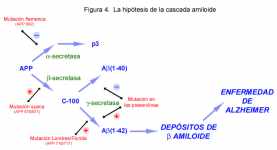
Figura 4 . La hipótesis de la cascada amiloide
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||