![]()
Enfermedad de Charcot-Marie-Tooth tipo 2
Dr. Onofre Combarros Pascual
Servicio de Neurología
Hospital "Marqués de Valdecilla"
39008, Santander
FAX: 202655
INTRODUCCION
Con el término de enfermedad de Charcot-Marie-Tooth (CMT) se designa al grupo más frecuente de neuropatías sensoriomotrices hereditarias cuya característica clínica es la presencia de déficit preferentemente motor y en menor grado sensitivo de distribución distal y carácter progresivo, y con un comienzo en los miembros inferiores y eventual posterior afectación de los superiores. La introducción en los años 60 y 70 de los estudios de conducción nerviosa y la práctica de biopsia de nervio periférico permitieron establecer dos tipos principales de CMT (Dyck y Lambert, 1968): 1/ la forma hipertrófica, desmielinizante o CMT tipo1, caracterizada por descenso de las velocidades de conducción nerviosa motora periférica , desmielinización de las fibras nerviosas mielínicas y crecimiento hipertrófico de las células de Schwann en "bulbos de cebolla"; 2/ la forma neuronal, axonal o CMT tipo 2, con velocidades de conducción nerviosa periférica normales o mínimamente descendidas y atrofia axonal primaria con mínima afectación de la mielina de las fibras nerviosas. Aunque en la mayoría de las series epidemiológicas la frecuencia encontrada para CMT2 es menor (una tercera parte) que para CMT1, nuestra experiencia en la población de Cantabria ha revelado que la prevalencia de ambos tipos es similar: 15.3 y 12.9 casos por 100.000 para CMT1 y CMT2, respectivamente (Combarros et al., 1987). CMT2 es genéticamente heterogénea porque existen varios posibles loci genéticos asociados a la enfermedad, y porque aunque la transmisión autosómica dominante es el modo principal de herencia, existen familias aisladas con transmisión autosómica recesiva (Harding y Thomas, 1980a).
CLINICA
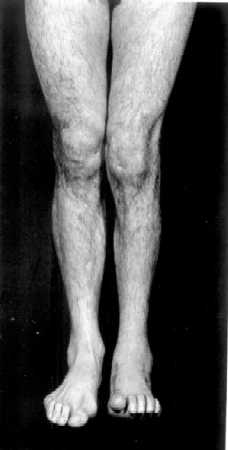
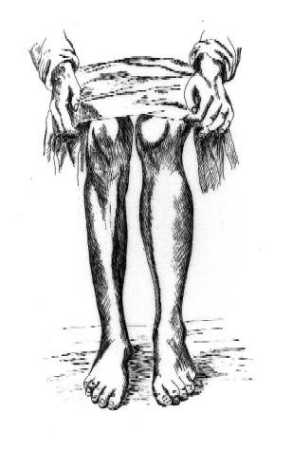
Atendiendo solo a las características clínicas, es imposible determinar la pertenencia de un paciente individual a las formas CMT1 o CMT2. Sin embargo, se han sugerido ciertas diferencias clínicas entre los grupos (Harding y Thomas, 1980b). Así, mientras que CMT1 tiene su edad de presentación en las dos primeras décadas, el rango de edades de comienzo de CMT2 es muy variable y se sitúa entre la segunda y la séptima décadas de la vida. Para un grado equivalente de debilidad muscular, la atrofia muscular parece ser más marcada en CMT2. En CMT2 los músculos de la pantorrilla sufren una afectación de igual intensidad a la de los del compartimento ánterolateral de la pierna, y no es raro encontrar clara asimetría en su distribución (Berciano et al., 1986), como puede observarse en la FIGURA 1 (obsérvese el paralelismo entre la asimetría de uno de nuestros pacientes y uno de los casos originales de la descripción de Tooth en 1886). Por el contrario, la afectación de la musculatura intrínsica de manos, el grado de arreflexia , la pérdida de la sensibilidad y la deformidad de los pies en cavo parecen ser más intensos en CMT1. Las raras formas autosómico recesivas (Harding y Thomas, 1980a; Ouvrier et al., 1981) son siempre más graves que las más habituales formas dominantes. Comienzan muy precozmente con retraso en la deambulación y tropiezos y caídas frecuentes. Muy pronto se aprecia un déficit motor distal en miembros inferiores y también en manos. La debilidad en manos es más pronunciada que en las formas dominantes. La presencia de dolor u otros síntomas sensitivos es rara, pero la exploración de la sensibilidad suele mostrar pérdida de la propiocepción. El pie cavo no es constante.
 |
 |
ELECTROFISIOLOGIA
El criterio fundamental que ha permitido la división de CMT en tipos 1 y 2 es el valor divisorio de la velocidad de conducción nerviosa motora (VCM) situado en 38 m/s (Dyck y Lambert, 1968; Harding y Thomas, 1980b). En todos los miembros afectos de familias con CMT2 existe una perfecta concordancia en sus valores de VCM que son ³ 38 m/s. Como es lo característico de una axonopatía distal, la normalidad o mínimo enlentecimiento de la VCM suele acompañarse de una caída en la amplitud de los potenciales motores en miembros inferiores, pero no siempre ocurre así en miembros superiores. Al estimular los nervios peroneal o mediano los potenciales motores solo están ausentes en aquellos casos con atrofia muscular marcada, y los músculos correspondientes muestran denervación y reinervación crónica en el EMG. En cambio, en pacientes asintomáticos las alteraciones electrofisiológicas pueden ser mínimas. Las alteraciones encontradas en los potenciales nerviosos sensitivos son superponibles a las de los motores. A diferencia de lo que ocurre con CMT1 en que los trastornos electrofisiológicos están precozmente presentes y permiten un rápido diagnóstico presintomático de los individuos en riesgo, en CMT2 estas anomalías suelen establecerse más tardíamente y progresan muy lentamente a lo largo del tiempo, y por ello el diagnóstico electrofisiológico de portadores de CMT2 puede ser difícil. Por otra parte, las alteraciones electrofisiológicas ya establecidas no son específicas, y si la historia familiar no es concluyente, pueden conducir al diagnóstico erróneo de neuropatía axonal adquirida (diabética o alcohólica) (Teunissen et al., 1997). Otro motivo de confusión diagnóstica acontece en familias sin transmisión varón-varón y que corresponden a CMT1 ligada al cromosoma X, al considerar como CMT2 a mujeres afectas o portadoras y que característicamente tienen alteraciones electrofisiológicas de "tipo axonal" diferentes a las de "tipo desmielinizante" encontradas en los varones afectos (Nicholson y Nash, 1993).
NEUROPATOLOGIA
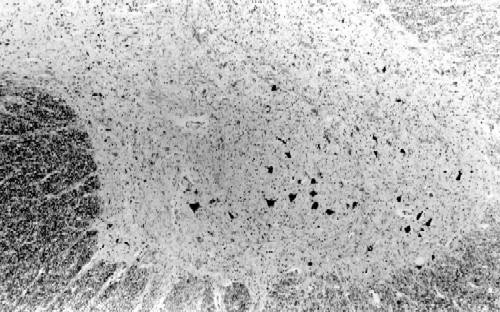
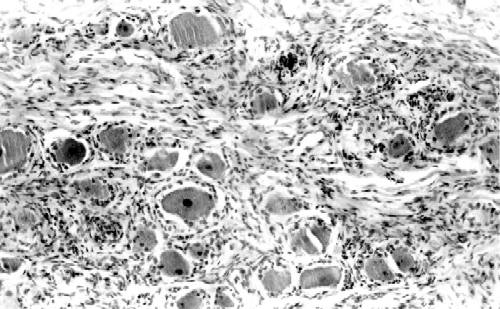
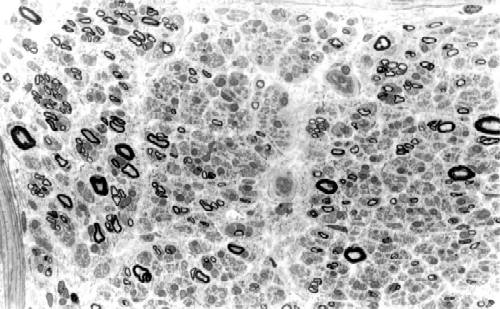
La biopsia de nervio sural demuestra pérdida de fibras mielínicas gruesas y clusters de regeneración con aumento del número de fibras mielínicas finas. Las fibras existentes tienen fenómenos de atrofia axonal y ocasionales acúmulos intraaxonales de neurofilamentos (Thomas et al, 1996), la presencia de desmielinización es muy rara y no se observan "bulbos de cebolla". En uno de los escasos estudios necrópsicos efectuados en CMT2 (Berciano et al., 1986), se observó una importante deplección de neuronas motoras de las astas anteriores lumbosacras (FIGURA 2) con degeneración axonal de las raíces anteriores, intensa pérdida de neuronas sensitivas en los ganglios de la raíz posterior (FIGURA 3) con degeneración axonal secundaria del fascículo gracilis y de las fibras mielínicas de las raíces posteriores, y pérdida de fibras mielínicas en los nervios periféricos (FIGURA 4) con una afectación distal mayor que proximal. Todos estos hallazgos son compatibles con atrofia neuronal primaria crónica con degeneración axonal distal.
GENETICA
Los primeros estudios de ligamiento genético descubrieron que CMT1 era genéticamente heterogénea y permitieron clasificarla en dos subgrupos principales: CMT1A con mutaciones en el gen PMP22 localizado en el cromosoma 17 (17p11.2) y CMT1B con mutaciones en el gen Po en el cromosoma 1 (1q22-q23). Bien pronto fue evidente que CMT2 no mapeaba en las regiones de estos dos genes de la mielina. La única excepción ha sido una gran familia italiana recientemente publicada (Marrosu et al., 1998), que cumplía los criterios exigibles para CMT2 y en la que los pacientes tenían una mutación puntual en el gen Po. Comoquiera que la expresión de este gen está restringida a la vaina de mielina y no al axón que es la diana lesional primaria en CMT2, este raro hallazgo necesita nueva confirmación. La primera evidencia de ligamiento genético en CMT2 fue publicada en 1993 (Ben Othmane et al), trás investigar seis grandes familias con herencia autosómico dominante y encontrar que en tres de ellas la enfermedad estaba asociada a marcadores del DNA situados en la región distal del brazo corto del cromosoma 1 (1p35-36). Así se estableció un primer subtipo denominado CMT2A. El cuadro clínico de los pacientes es el habitual en CMT2 (Saito et al., 1997). Este primer locus es, sin embargo, poco frecuente porque de 11 posteriores familias estudiadas solo una mostró ligamiento a este locus (Timmerman et al., 1996). Un segundo locus genético para CMT2 (CMT2B) fue localizado en el cromosoma 3 (3q13-22) en una familia cuyo fenotipo se distinguía por la importancia de las alteraciones de la sensibilidad presentes. Así, junto a la existencia de pie cavo y debilidad y amiotrofia distal en miembros, existía una pérdida importante de la sensibilidad en los pies con desarrollo de úlceras plantares, sin dolor ni parestesias y sin disautonomía asociada (Kwon et al., 1995). Este fenotipo con acropatía úlceromutilante que recuerda a la neuropatía sensitiva hereditaria tipo I ha sido observado de nuevo en otra familia con CMT2B (De Jonghe et al., 1997). El subgrupo CMT2C engloba a dos familias descritas por Dyck et al (1994), y que pese a carecer de ligamiento genético conocido se distinguen clínicamente por la presencia de debilidad de cuerdas vocales y diafragma. El último locus conocido (CMT2D) está situado en el cromosoma 7 (7p14)) y ha sido hallado en una familia dominante caracterizada porque la atrofia y debilidad muscular comienzan y son más grave en las manos que en los pies (Ionasescu et al., 1996). El siguiente paso en la investigación genética ha de ser el descubrir nuevos loci genéticos ya que la mayor parte de las familias CMT2 no se ligan a ninguno de los tres hasta ahora descritos (Timmerman et al., 1996), y también, es necesario identificar los genes responsables de CMT2 situados en estos loci conocidos. Sin duda éste es el inicio del largo camino hacia la consecución de una terapia génica para este proceso.
BIBLIOGRAFIA
Ben Othmane K, Middleton LT, Loprest LJ, et al (1993). Localization of a gene (CMT2A) for autosomal dominant Charcot-Marie-Tooth disease type 2 to chromosome 1p and evidence of genetic heterogeneity. Genomics 17: 370-375.
Berciano J, Combarros O, Figols J, et al (1986). Hereditary motor and sensory neuropathy type II. Clinicopathological study of a family. Brain 109: 897-914.
Combarros O, Calleja J, Polo JM, Berciano J (1987). Prevalence of hereditary motor and sensory neuropathy in Cantabria. Acta Neurol Scand 75: 9-12.
De Jonghe P, Timmermann V, FitzPatrick D, Spoelders P, Martin JJ, Van Broeckhoven C (1997). Mutilating neuropathic ulcerations in a chromosome 3q13-q22 linked Charcot-Marie-Tooth disease type 2B family. J Neurol Neurosurg Psychiatry 62: 570-573.
Dyck PJ, Lambert EH (1968). Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol 18: 603-618.
Dyck PJ, Litchy WJ, Minnerath S, et al (1994). Hereditary motor and sensory neuropathy with diaphragm and vocal cord paresis. Ann Neurol 35: 608-615.
Harding AE, Thomas PK (1980a). Autosomal recessive forms of hereditary motor and sensory neuropathy. J Neurol Neurosurg Psychiatry 43: 669-678.
Harding AE, Thomas PK (1980b). The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 103: 259-280.
Ionasescu V, Searby C, Sheffield VC, Roklina T, Nishimura D, Ionasescu R (1996). Autosomal dominant Charcot-Marie-Tooth axonal neuropathy mapped on chromosome 7p (CMT2D). Hum Mol Genet 5: 1373-1375.
Kwon JM, Elliott JL, Yee W (1995). Assignement of a second Charcot-Marie-Tooth type II locus to chromosome 3q. Am J Hum Genet 57: 853-858.
Marrosu MG, Vaccargiu S, Marrosu G, Vannelli A, Cianchetti C, Muntoni F (1998). Charcot-Marie-Tooth disease type 2 associated with mutation of the myelin protein zero gene. Neurology 50: 1397-1401.
Nicholson G, Nash J (1993). Intermediate nerve conduction velocities define X-linked Charcot-Marie-Tooth neuropathy families. Neurology 43: 2558-2564.
Ouvrier RA, McLeod JG, Morgan GF, Wise GA, Conchin TE(1981). Hereditary motor and sensory neuropathy of neuronal type with onset in early childhood. J Neurol Sci 51: 181-197.
Saito M, Hayashi Y, Suzuki T, Tanaka H, Hozumi I, Tsuji S (1997). Linkage mapping of the gene for Charcot-Marie-Tooth disease type 2 to chromosome 1p (CMT2A) and the clinical features of CMT2A (1997). Neurology 49: 1630-1635.
Teunissen LL, Notermans NC, Franssen H, et al (1997). Differences between hereditary motor and sensory neuropathy type 2 and chronic idiopathic axonal neuropathy. A clinical and electrophysiological study. Brain 120: 955-962.
Thomas PK, King RHM, Small JR, Robertson AM (1996). The pathology of Charcot-Marie-Tooth disease and related disorders. Neuropathol Appl Neurobiol 22: 269-284.
Timmermann V, De Jonghe P, Spoelders P, et al (1996). Linkage and mutation analysis of Charcot-Marie-Tooth neuropathy type 2 families with chromosomes 1p35-p36 and Xq13. Neurology 46: 1311-1318.