![]()
GENETICA DE LA ENFERMEDAD DE CHARCOT-MARIE-TOOTH TIPO 1, DE LAS NEUROPATIAS FOCALES HEREDITARIAS Y DE LAS NEUROPATIAS HEREDITARIAS MOTORAS DISTALES
Teresa Sevilla
Hospital Universitari La Fe de Valencia. España.
Resumen
La enfermedad de Charcot-Marie-Tooth (CMT) o neuropatía hereditaria motora y sensitiva (NHMS) presenta una gran heterogeneidad genética. Las formas tipo 1 (CMT 1) o desmielinizantes, tipo 2 (CMT 2) o neuronales son dos formas clínicas y genéticas distintas. Además, aparte de las formas motoras y sensitivas previamente citadas existe otro tipo de neuropatía con afectación clinica y neurofisiológica exclusivamente motora denominada atrofia espinal distal (AED) o neuropatía motora hereditaria distal (NMHD) que sindrómicamente se encuadra dentro de la enfermedad de Charcot-Marie-Tooth. La CMT 1 es la forma más prevalente y la más conocida desde el punto de vista genético. Se han descrito al menos cuatro genes involucrados en la misma: el gen de la proteína mielínica períferica de 22 kDa (PMP22), localizado en el cromosoma 17p11.2 (locus CMT1A), el gen de la proteína 0 de la mielina (P0), localizado en el cromosoma 1q23 (locus CMT1B), el gen de la conexina 32 (Cx32), localizado en el cromosoma Xq13 (locus CMTX) y el gen EGR2 localizado en el cromosoma 10q21.1-q22.1. Los genes PMP-22 y P0 se asocian a formas autosómico dominantes, el gen Cx32 se transmite ligado al cromosoma X y el gen EGR2 se ha demostrado su transmisión como forma autosómico dominante y autosómico recesiva. En las formas desmielinizantes con herencia autosómico recesiva se ha hallado ligamiento positivo en cuatro locus diferentes: 8q 21.1 (CMT4A), 11q23 (CMT4B), 5q23-33 (CMT4C) y 8q24 en una forma asociada a sordera (CMT-Lom).
Para las formas autosómico dominantes de las NMHD se han identificado dos locus: una gran familia de inicio y predominio en extremidades superiores que mapea en el cromosoma 7p (NHMD V), y otra con el fenotipo clásico que mapea en el cromosoma 12q24.
En la neuropatía familiar con susceptibilidad a las parálisis por presión (NFSP) se ha encontrado en la mayoría de las familias deleción del locus CMT1A, y en raros casos mutación puntual del gen PMP-22. En la neuralgia amiotófica familiar el análisis de genética molecular ha mostrado ligamiento con marcadores en el cromosoma 17q24-25
HISTORIA
Hace más de un siglo que Charcot y Marie en Francia (1) y Tooth en Inglaterra (2) describieran con detalle una nueva forma de atrofia muscular progresiva que denominaron "atrofia muscular peroneal", lo que hoy conocemos por síndrome de Charcot-Marie-Tooth (CMT). Las enfermedad tenía un caracter familiar, los síntomas comenzaban en la infancia con debilidad y atrofia de la musculatura peroneal y la progresión era lenta. Pocos años después Dejerine y Sottas describieron una neuropatía en dos hermanos, en la hermana los síntomas debutaron en la infancia con retraso del inicio de la marcha a los tres años y con el tiempo desarrolló importantes atrofias distales y alteraciones sensitivas de la coordinación; en el hermano la enfermedad comenzó a los 14 años y el curso fue menos grave. Los nervios estaban hipertróficos (3). En 1926 Roussy y Lévy publicaron las características clínicas de siete miembros de una familia con veinte afectos; el síndrome clínico era semejante al descrito por Charcot, Marie y Tooth, salvo que la atrofia no existía o era mínima, la sensibilidad era normal y cuatro pacientes mostraban temblor al ejecutar movimientos (4). Lapresle y Salisachs demostraron en un caso de esta familia que las características neurofisiológicas y patológicas eran idénticas a la enfermedad de Charcot-Marie-Tooth (5).
En los años sesenta Dyck y Lambert a partir de un estudio prospectivo de familias con neuropatías hereditarias clasificaron a los pacientes en dos grandes grupos según la velocidad de conducción nerviosa y la biopsia de nervio sural. El tipo 1 (CMT1) o neuropatía hereditaria motora y sensitiva tipo 1 (NHMS I) cursaba con velocidad de conducción enlentecida y en la biopsia se encontró desmielinización segmentaria y en algunos casos hipertrofia; en el tipo 2 (CMT2) o neuropatía hereditaria motora y sensitiva tipo 2 (NHMS II) la velocidad de conducción estaba normal o relativamente conservada y los hallazgos de la biopsia eran compatibles con una neuropatía axonal. Los casos de herencia recesiva o esporádicos, semejantes al tipo I pero más agresivos los denominaron NHMS tipo III o enfermedad de Dejerine-Sottas (6,7). Aunque posteriormente varios investigadores reafirmaron esta subdivisión genérica (8-14), no fue unívocamente aceptada. Así, Humberstone y Salisachs encontraron una distribución aleatoria de las velocidades de conducción (15,16) y Davis y cols. identificaron un grupo con velocidad de conducción intermedia (17).
En los últimos 30 años ha habido numerosas publicaciones de neuropatías con inicio de la enfermedad al nacimiento con hipotonía marcada, llanto débil y debilidad para tragar y respirar. En estos niños el desarrollo motor estaba considerablente enlentecido y algunos fallecieron en la infancia por insuficiencia respiratoria y neumonía por aspiración. La biopsia de nervio mostró ausencia virtual de las vainas de mielina (18-27)
HETEROGENEIDAD GENETICA
La presencia de heterogeneidad que se había puesto de manifiesto con los estudios electrofisiológicos y morfológicos se ha corroborado en la última década con la tecnologia del DNA recombinante. Bird y cols. encontraron ligamiento significativo en la forma desmielinizante de la enfermedad en un locus del cromosoma 1 próximo al antígeno Duffy de los hematíes (28), este hallazgo no se constató en la forma neuronal corroborando así su independencia genética (29). También se encontró ausencia de correlación de este locus en otra larga familia con una forma desmielinizante (30). Estos fueron los primeros descubrimientos que vislumbraron la presencia de heterogeneidad en el síndrome de Charcot-Marie-Tooth, desde entonces hasta hoy se han identificado varios genes candidatos y numerosos locus han sido asignados. De este modo la clasificación se va haciendo cada vez más abigarrada y resulta dificil recordar y correlacionar tipo y alteración genética subyacente. Por si esto fuera poco todavia se tiende a relacionar los síndromes clásicos (vgr. Dejérine-Sottas, Roussy-Levi) con determinados tipos de herencia o gravedad clínica. En general y a efectos didácticos creo que deberíamos dividir el síndrome CMT en dos grandes grupos: CMT1 y CMT2. A partir de aquí cualquiera de los dos puede tener patrón de herencia autosómico dominante, autosómico recesiva o casos esporádicos. Las familias con patrón de herencia ligado al cromosoma X merecerán una mención aparte por la peculiaridad de las mismas y la dificil adjudicación a los tipos CMT1 o CMT2.
FENOTIPOS
Aunque existen gran confusión en cuanto a la terminología y toponimias de los diferentes síndromes clinicos y no se ha establecido aún ningún consenso, intentaré definir lo que el sentir de la mayoría de los autores interpreta por los siguientes términos.
Fenotipo CMT1: características clínicas del fenotipo originalmente descrito por Charcot, Marie y Tooth. Las características incluyen debilidad distal y atrofia de inicio en musculatura peroneal con eventual extensión a dedos de las manos, hipo o arreflexia, pies cavos, engrosamiento de los nervios a la palpación en algunos casos y alteraciones sensitivas distales discretas al examen clínico. El grado de afectación es leve o moderado y los paciente mantienen la independencia incluso en la vejez. Electrofisiológicamente la velocidad de conducción motora está enlentecida con un valor de corte en el nervio mediano menor de 38 m/s. La biopsia de nervio sural muestra signos de desmielinización y remielinización con la formación de "bulbos de cebolla" que se corresponden a engrosamientos concéntricos de las prolongaciones de las células de Schwann ( 31 ).
Fenotipo CMT2: la clínica es semejante al CMT1 aunque la edad de inicio suele ser algo más tardía y la musculatura intrínseca de las manos se afecta en menos casos. La diferencia fundamental es que la velocidad de conducción motora es normal o casi normal y la biopsia de nervio sural muestra signos de degeneración axonal con preservación de las vainas de mielina.
Fenotipo síndrome de Dejerine-Sottas: generalmente se suele utilizar para casos de neuropatía grave, de comienzo en la infancia y con gran enlentecimiento de la velocidad de conducción. Suelen ser casos esporádicos o herencia recesiva. Los nervios pueden estar engrosados y en la biopsia se observa importante pérdida de fibras mielinizadas y numerosos bulbos de cebolla.
Fenotipo neuropatía hipomielinizante congénita: el inicio es al nacimiento con hipotonía, llanto débil y debilidad para respirar y tragar. La diferencia fundamental estriba en la biopsia, en la que se encuentra ausencia virtual de vainas de mielina.
NEUROPATIAS DE TIPO DESMIELINIZANTE
Enfermedad de CMT1 autosómico dominante
CMT1A
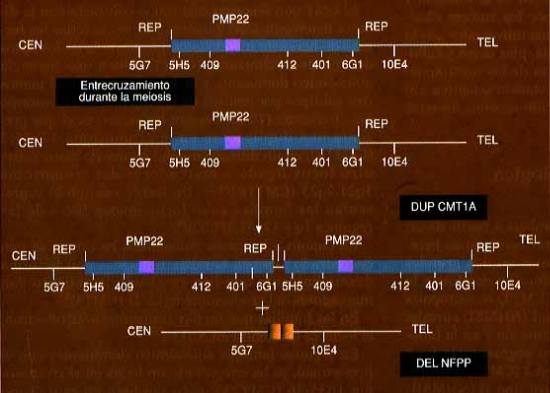
La mayoría de las familias con enfermedad de CMT1A presentan una duplicación en el segmento 17p11.2 que segrega con la enfermedad y abarca un tamaño aproximado de 1500 kilobases (fig.1) (32-34). Esta región duplicada se dispone en tandem y está flanqueada por secuencias repetitivas, denominadas CMT1A-REP, que mapean proximales y distales a la duplicación CMT1A (35,36). Las secuencias CMT1A-REP están presentes en tres copias en el caso de la duplicación y en una en el caso de que exista deleción como se ha desmostrado en otra neuropatía desmielinizante y familiar que cursa con episodisos recurrentes de parálisis en los nervios más propensos a sufrir compresiones o traumatismos (36). Así pues, dos neuropatías hereditarias diferentes podrían ser explicadas por un único mecanismo: un entrecuzamiento desigual en la meiosis entre cromosomas homólogos daría como resultado una duplicación de un cromosoma y la deleción en el homólogo (fig. 2). La duplicación se ha detectado en familias CMT1A de origen étnico y geográfico diferente (33, 37-40).
No siempre la enfermedad CMT1A está debida a una duplicación, en un pequeño número de casos se debe a una mutación puntual en el mismo locus (41-46).
El PMP-22 es un gen candidato para la CMT1A
El descubrimiento de un ratón llamado Trembler (Tr), que sufría crisis y temblores y que a nivel celular tenía hipomielinización del sistema nervioso periférico y proliferación de las células de Schwann proporcionó el modelo murino de la enfermedad CMT. Se describieron dos mutaciones puntuales en el gen para la proteína-22 de la mielina (PMP-22) localizado en el cromosoma 11 murino en una región que se correponde con el cromosoma 17 humano, lo que hizo pensar que el gen PMP-22 humano podría encontrarse en el brazo corto del cromosoma 17 (47,48). El PMP-22 se clonó y se demostró que codifica un polipéptido de 160 aminoácidos, idéntico en longitud al que previamente se había clonado en el ratón. (49). Una vez clonado el gen, se determinó que su localización estaba en la región duplicada del cromosoma 17 (17p11.2-17p12) y en doble dosis en los cromosomas duplicados, lo que lo convertía como un importante candidato para la CMT1A (49-51). Además, se pudo demostrar que el gen PMP-22 humano se expresaba fundamentalmente en las células de Schwann (determinando la producción de RNA en varios tejidos humanos), resultados idénticos a los estudios previos de PMP-22 en ratón (52).
Dos razones principales avalan al gen PMP-22 como candidato para la enfermedad CMT1A: su localización dentro de la región duplicada y su expresión principal en nervio periférico.
¿ Por qué mecanismo el PMP-22 produce un fenotipo CMT1A ?
Aunque varios mecanismos podrían explicar porqué el gen PMP-22 produce el fenotipo CMT1A la evidencia apoya que el efecto de dosis (tres copias en lugar de dos) es la explicación más atractiva. Este mecanismo lo avalan varios hechos: 1) el haber encontrado un fenotipo más grave en un paciente con expresión homocigota (4 copias), 2) el que existan pacientes con trisomías parciales del cromosoma 17p y enlentecimiento de la velocidad de conducción y 3) haber hallado casos con una duplicación de menor tamaño que la habitual pero que contienen 2 copias del gen (53).
Por otra parte también se ha descrito familias con enfermedad CMT1A que no presentaban la duplicación pero que tenían mutaciones puntuales en el PMP-22 (41-46).
El hallazgo de mutaciones puntuales nos plantea importantes cuestiones con respecto a la patogenia de la enfermedad y reconcilia las hipótesis de sobre-expresión de un gen y sustitución de un aminoácido como mecanismos etiológicos subyacentes. La elucidación de la función biológica del PMP-22 es crítica para desenmarañar los mecanismos exactos que ayuden a comprender como una duplicación y un mutación puntual llevan al mismo fenotipo clínico y electrofisiológico. Entender la fisiopatología básica puede llevar al descubrimiento de terapias para la enfermedad de CMT y otras neuropatías hereditarias.
En el Hospital la Fe de Valencia hemos analizado la presencia de la duplicación en 17p11.2 en 26 familias autosómico dominantes y la duplicación se encontró en el 92% de las mismas.
PMP-22 Y OTROS FENOTIPOS
Se han identificado mutaciones puntuales de-novo en pacientes con fenotipo Dejerine-Sottas (54,55). En estos casos la mutación se ha presentado en estado heterocigoto, lo que sugiere un efecto autosómico dominante. Nosotros hemos hallado una mutación puntual en el exón 3 del gen PMP22 (substitución de una serina por una prolina en la posición 79) en una niña con un fenotipo muy grave que se presenta en estado heterocigoto (56) . Los datos de genética molecular apoyan que los síndromes CMT y DS representan un espectro de fenotipos clínicos que pueden surgir por diferentes mutaciones en un mismo gen.
En la Neuropatía familiar con susceptibilidad a las parálisis por presión (NFSP) se ha encontrado deleción de 1.5 MB del fragmento 17p11.2 en algunas familias (57), y en otras que no tenían deleción del citado fragmento se halló una deleción de dos pares de bases (58) lo que aporta evidencia del importante papel del PMP22 en la enfermedad.
La mayoría de los casos esporádicos con fenotipo CMT1 presentan la duplicación en 17p11.2, lo quiere decir que estamos ante una mutación de novo, ausente en los padres y que a partir de la cual se transmitirá a la descendencia con la misma proporción que los casos dominantes (59). En la mayoria de los casos (89%) la duplicación es de origen paterno y se debe a un entrecruzamiento desigual entre cromátides no hermanas en la meiosis, este intercambio daría como resultado dos mutaciones recíprocas, una duplicación en un cromosoma y una deleción en el homólogo (60). Como hemos comentado previamente la deleción se correponde con la NFSP. En un reducido porcentaje de casos esporádicos la duplicación puede ser de origen materno (11%) por rearreglo intracromosómico: desigual intercambio entre cromátides hermanas (61).
CMT1B
En 1980 Bird y cols. encontraron ligamiento entre el antígeno Duffy de los hematies y el síndrome CMT1 (29), se designó como enfermedad CMT1A a las familias no ligadas al antígeno Duffy y CMT1B a las familias ligadas al mismo (31). Aunque este fué el primer locus hallado para la enfermedad CMT1 el gen candidato no se encontró hasta 1993 tras la caracterización y clonación del gen de la proteína cero (P0) de la mielina cuya localización coincidia con el locus CMT1B (62).
La P0 es la proteína más abundante de la mielina del sistema nervioso periférico y tiene un papel importante en hacerla compacta (63). Tras la caracterización del gen P0 se han encontrado varias mutaciones puntuales en determinadas familias CMT1B que segregan con la enfermedad. También se ha encontrado deleción de un codón en una familia (64) así como mutaciones de novo en determinados en pacientes descritos como fenotipo Dejerine-Sottas (65-69). Las mutaciones en el gen que codifica la P0 son heterocigotas y presumiblemente expresan la mitad de la dosis de la proteína normal (efecto dominante-negativo). Animales homocigotos con una mutación nula en el gen P0 muestran una neuropatía grave desde los primeros días con descompactación de las vainas de mielina y posterior formación de bulbos de cebolla, los animales heterocigotos están mucho menos afectos con formación normal de la mielina en los jóvenes, seguida por una progresiva desmielinización en los más adultos (70). Se han encontrado múltiples mutaciones puntuales que figuran en la tabla 2.
CMT1C
Se han publicado al menos dos familias que no mostraron ligamiento a los cromosomas 1 ni 17, sugiriendo la posibilidad de un tercer locus para la enfermedad CMT1 autosómica dominante y ampliando el espectro de la heterogeneidad (71).
Enfermedad de CMT autosómico recesiva
La herencia autosómico recesiva no debe asumirse a menos que ambos padres hayan sido examinados clínicamente y, preferiblemente con estudios neurofisiológicos, ya que los signos clínicos de la enfermedad pueden ser muy leves. La realización de estudios genéticos en las formas autosómico recesivas es mucho más complicado que en las dominantes, debido a la escasez de familias que la presentan y generalmente a que las fratrias suelen ser muy cortas. Hasta ahora se han descubierto cuatro locus que pasaré a detallar a continuación. La terminologia de las formas autosómico recesivas de las neuropatias genéticas desmielinizantes es un poco confusa ya que clásicamente se solia denominar síndrome de Dejerine-Sottas o neuropatía hereditaria sensitivomotora tipo III (NHSM III). El primer locus recesivo se asignó a un grupo de familias tunecinas y se le designó como CMT4 tipo A (CMT4A) (72), posteriormente la mayoría de las formas recesivas se conocerán como CMT4 y la correspondiente letra alfabética.
CMT4A (forma Tunecina)
La primera evidencia de ligamiento significativo fué hallada en una serie de 4 familias tunecinas consanguineas con neuropatía desmielinizante (72). Los marcadores del ligamiento estaban en el cromosoma 8q-21.1 y no se encontró evidencia de heterogeneidad genética en las familias. Este grupo de familias era clínicamente homogéneo y se caracterizaba por una edad de comienzo precoz y debilidad distal grave. Hasta el momento actual se desconocen genes candidatos en esta región.
CMT4B (Neuropatia con desdoblamientos focales de mielina)
A este segundo grupo pertenecen una serie de pacientes con neuropatia autosómico recesiva que en la biopsia se caracterizan por engrosamientos o desdoblamientos focales de la vaina de mielina. El locus para una larga familia con un alto índice de consanguinidad se ha hallado en el cromosoma 11q23 (73). Otra familia corta, con solo 2 afectos y clínica y patologia superponible a la ligada al 11q23 no mostró ligamiento positivo para este locus ni ningún otro conocido (74).
CMT4C (forma clásica)
Un tercer locus se ha encontrado en dos largas familias argelinas consanguíneas, cuyas manifestaciones clínicas, electrofisiologicas y neuropatológicas semejan a las formas autosómico dominantes de CMT1. Este locus se ha asignado al cromosoma 5q23-33 (75).
CMT-Lom (asociada a sordera)
Este tipo de neuropatía desmielinizante se ha hallado en una comunidad gitana de Bulgaria, aunque no se encontraron matrimonios consanguíneos el grupo es altamente endogámico. En este caso la neuropatía se asocia a sordera que suele comenzar alrededor de la tercera década (76). Los marcadores genéticos han sido significativos para un locus localizado en el cromosoma 8q24 (77).
Enfermedad de CMT ligada al cromosoma X
La herencia ligada al cromosoma X se deduce observando el árbol genealógico y viendo que no existe transmisión hombre-hombre. La existencia de esta forma de herencia fue sugerida por Allan (78). Actualmente se le suele encuadrar bajo el encabezamiento de CMT1 pero en sentido estricto es díficil aceptar esta clasificación ya que no todos los miembros de la familia tienen una velocidad de conducción motora <38 m/s ni la biopsia de nervio es superponible a la enfermedad CMT1. El patrón clínico es bastante uniforme en las diferentes familias, los hombres experimentan síntomas en la primera década y el grado de afectación suele ser bastante importante; las mujeres no suelen tener síntomas hasta el final de la segunda década o más adelante y en general muestran escasos signos de enfermedad (79-83). La velocidad de conducción motora (VCM) varia entre los diversos individuos de una familia, los más gravemente afectos tienen VCM enlentecida y los ligeramente afectos la tienen normal o casi normal (84). En cuanto a la patologia también difiere de la CMT1 ya que se encuentra mayor densidad de fibras mielinizadas, mayor número de fibras regenerantes ("clusters") y pocos bulbos de cebolla (85). El gen para la CMTX-L mapea en Xq13.1 y codifica la proteína Conexina-32 (86,87), que se localiza adyacente a a los nodos de Ranvier y las incisuras de Smidt-Lanterman. Subunidades de esta proteína se ensamblan para formar canales que interactuan con otros canales en las células adyacentes para el transporte de iones y pequeñas moléculas . Se han identificado mutaciones puntuales, deleciones de un codón e inserciones de pares de bases (88-91). El análisis mutacional de la Cx-32 en pacientes con enfermedad de CMT ha demostrado que las familias con esta mutación son mucho más frecuentes que lo que previamente se hubiera esperado. La CMT con mutación de la Cx-32 es la segunda más prevalente precedida por la CMT1A con duplicación.
Otros genes asociados con neuropatias genéticas desmielinizantes
EGR2
¿Que es?
Es parte de una familia de multigenes que que codifica proteínas que tienen que ver con la regulación de la proliferación celular. El locus está en el cromosoma 10q21.1-q22.1 (92).
A pesar de que se conocen mutaciones en al menos 3 genes que intervienen en el proceso de regulación de la mielelinización del sistema nervioso periférico (PMP-22, MPZ y Cx-32 ) aún hay casos en los que aún no se ha descubierto alteración en ninguno de estos genes conocidos. Esto sugiere que probablemente existan otros genes que se requieran para la formación, mantenimiento y compactación de la mielina, que bien se encuentren en alguno de los locus descritos o en otros aún no conocidos. Basados en observaciones en animales en que el ratón que carece del gen EGR2 (Egr2 -/-) muestra un fenotipo con hipomielinización de los nervios periféricos y bloqueo de la diferenciación de las células de Schwann se sugiere que el gen EGR2 puede ser un buen candidato para el control de la mielinización en el SNP. Según esta hipótesis Warner y cols. han identificado una mutación en una familia con fenotipo CMT1 autosómico dominante (en un alelo), otra mutación recesiva (en los 2 alelos) en tres pacientes con neuropatía cogénita hipomielinizante (NCH) autosómico recesiva y una mutación dominante de novo en un caso esporádico de NCH. Se descartó la posibilidad de polimorfismo en 100 controles (92). Se puede sugerir que diferentes mutaciones del mismo gen podrían dar por resultado detención del proceso de mielinización (NCH) y desmielinización (CMT1).
La asociación de mutaciones en un gen que potencialmente activa otros genes necesarios para la mielinización del SNP apoya la hipótesis que las neuropatías genéticas desmielinizantes o hipomielinizantes pueden resultar de diferentes alteraciones en la estructura o formación de la mielina (92).
Neuropatia congénita hipomielinizante
Este término se utiliza practicamente como sinónimo de sindrome de Dejerine-Sottas, la diferencia solo podría definirse tras estudiar la patologia demostrando que apenas hay fibras mielinizadas en la biopsia de nervio. El comienzo de la enfermedad es desde el nacimiento con hipotonia, ausencia de los reflejos neonatales, retraso de las adquisiciones motoras y a veces distress respiratorio. Mutaciones en casos con NCH se han descrito en el gen PMP22 en estado heterocigoto (56), en el gen EGR2 en estado heterocigoto y homocigoto (92) y en el gen MPZ en estado heterocigoto (93).
La interrelación entre mutaciones de los diferentes genes y los fenotipos hallados se muestra en la tabla 2.
NEUROPATIAS FOCALES HEREDITARIAS
Neuropatia familiar con susceptibilidad a la presión
La neuropatia familiar hereditaria con susceptibilidad a la presión (NFSP) o polineuropatía recurrente familiar, es una enfermedad autosómico dominante que produce de forma episódica una neuropatía recurrente y desmielinizante (94). De Jong fue el primero en describir detalladamente la clínica de la enfermedad (95). La NFSP suele comenzar en la adolescencia y puede causar ataques de parestesias, debilidad muscular y atrofia. En la mayoría de las ocasiones la parálisis desaparece en días, semanas o meses aunque algunas veces la incapacidad puede ser de larga duración o permanente (94, 96, 97). En algunos casos, la clínica suele desencadenarse tras un mínimo trauma en los nervios periféricos. El síndrome del tunel carpiano y otras neuropatías por atrapamiento son manifestaciones frecuentes de esta enfermedad. La velocidad de conducción motora y sensitiva suele estar algo disminuida en los afectos y portadores asintomáticos. Los cambios patológicos incluyen desmielinización segmentaria y engrosamiento de algunos segmentos de la vaina de mielina ("salchichas").
Es importante hacer el diagnóstico diferencial con otra neuropatía familiar recurrente que es la neuralgia amiotrófica hereditaria (NAH) (98,99) ya que en la PHPC no es rara la afectación del plexo braquial (100). Una diferencia importante es que el dolor y la atrofia muscular son importantes en la plexopatía braquial y no se menciona en la PHNC (94,98).
Chance y cols. descubieron que en algunas familias con NFSP la alteración genética que subyace es una deleción en el cromosoma 17p11.2 (57) que incluye todos los marcadores que están duplicados en el CMT1A y el gen PMP-22.( fig 1 y 2), otros investigadores corroboraron este hallazgo (101-105) Además de la deleción existen familias con fenotipo NFSP y mutación puntual del PMP-22 (58, 106,107) lo que pone de manifiesto la importancia del papel de este gen en la fisiopatologia de las neuropatías desmielinizantes hereditarias. La prevalencia de la deleción en una serie fue del 66% (108 ).
La demostración de una deleción en el cromosoma 17p11.2 en la NFSP extiende la descripción de los distintos fenotipos que aparentemente resultan por diferencias en el número de copias genéticas en esta región. La monosomía por deleción está asociada con NFSP mientras que la trisomía por duplicación resulta en CMT1A. Estas dos enfermedades pueden constituir un modelo importante para el estudio de las consecuencias fenotípicas que resultan de la variación de dosis génica. En la NFSP se ha demostrado que la cantidad relativa de PMP-22 mRNA en los nervios surales de pacientes afectos con respecto a controles estaba significativamente disminuida, lo que apoya que una disminución de la dosis del gen sea el mecanismo patogénico más probable. (109). Además tambien se ha demostrado una correlación entre la disminución de la expresión del mRNA del PMP-22 y la gravedad de la enfermedad (110).
Neuralgia amiotrófica hereditaria
La neuralgia amiotrófica hereditaria con predilección por el plexo braquial es una enfermedad autosómico dominante que cursa con ataques de dolor, debilidad muscular, y alteraciones sensitivas en la región braquial y que habitualmente comienza en la infancia. La recuperación parcial o total dura entre 1-3 meses. En muchas familias se han asociado características dismórficas, incluyendo talla corta, hipotelorismo, paladar hendido, epicanto, asimetría facial, y sindactilia parcial (111). Los estudios neurofisiológicos muestran una interrupción axonal a nivel del plexo braquial en las extremidades afectas, las velocidades de conducción motora son normales. La patologia es compatible con degeneración axonal.
No se conoce aún el gen causante de la enfermedad pero los analisis de genética molecular han mostrado ligamiento con marcadores en el cromosoma 17q24-25 (112).
NEUROPATIAS MOTORAS HEREDITARIAS DISTALES (NMHD)
Dentro del síndrome de Charcot-Marie-Tooth existe un tipo de afectación que cursa solo con debilidad muscular y atrofia sin afectar a la sensibilidad, clínica ni electrofisiológicamente. Dyck y Lambert denominaron a esta enfermedad "forma espinal de la enfermedad de Charcot-Marie-Tooth o atrofia espinal distal" (7). Los estudios neurofisiológicos muestran velocidad de conducción motora normal y ausencia de afectación de los nervios sensitivos; la biopsia de nervio sural es normal. En general la enfermedad se inicia en extremidades inferiores pero como variantes cabe citar a algunas familias en las que la enfermedad es de inicio y predominio en extremidades superiores (113), y se han descrito casos familiares que asociaron a la amiotrofia distal una paresia de las cuerdas vocales (114-117) .
En cuanto a la frecuencia, en la mayoria de las series publicadas los casos de NMHD están incluidos en las series del sindrome de atrofia peroneal y los rangos de frecuencia respecto a las neuropatias hereditarias motoras y sensitivas oscilan entre el 3 y el 6%. En la serie de Harding y Thomas (118) la frecuencia respecto a las NHMS fue del 12,5% y en la nuestra de12,28% (119).
La NMHD es clínica y genéticamente heterogénea y se ha clasificado en 7 subtipos de acuerdo a la edad de inicio, evolución y patrón de herencia (120,121). Hoy día se conocen al menos 2 locus implicados.
Neuropatia hereditaria motora distal tipo II
Timmerman y cols. realizaron un estudio clínico-genético de una familia con neuropatia hereditaria motora distal de inicio en el adulto y de herencia autosómico dominante (NHMD II) con seis generaciones. El ligamiento fue significativo para marcadores localizados en el cromosoma 12q24 (122). Varios genes se encuentran localizados en esta región, entre ellos los de la fosfolipasa A2, el de la oxido nítrico sintetasa y otro gen que se expresa en los tumores neuroendocrinos y podría actuar como regulador de la diferenciación celular.
Neuropatia hereditaria motora distal NHMD tipo V
Este locus fue el primero que se asignó para una neuropatía motora distal y se localizó en brazo corto del cromosoma 7. La clínica comenzo con una media de edad de 17 años y la amiotrofia se inició en extremidades superiores, afectando posteriormente a los inferiores en el 40% de los miembros de la familia, la progresión de la enfermedad ha sido muy lenta en esta familia (123). El tipo de herencia es autosómico dominante.
Tabla 1. Clasificación del síndrome de Charcot-Marie-Tooth
- Autosómico dominante
- CMT 1A duplicación 17p11.2
- CMT 1B mutación del gen P0 de la mielina
- CMT 1C no A / no B
- Mutación EGR2
- Autosómico recesiva
- CMT 4A ligada al cr. 8q13-q21.1 (forma Tunecina)
- CMT 4B ligada al cr. 11q23 (con desdoblamientos de la mielina)
- CMT 4C ligada al cr. 5q23-q33 (forma clásica)
- CMT-Lom ligada al cr. 8q24 (asociada a sordera)
- Autosómico dominante
- CMT 2A ligada al cr. 1p35-36
- CMT 2B ligada al cr. 3q13-22
- CMT 2C asociada a paresia de cuerdad vocales
- CMT 2D ligada al cr. 7p14
- Autosómico recesiva (no locus)
- Mutaciones de la Conexina-32
- Mutaciones puntuales PMP-22, P0 y EGR2
- Neuropatía familiar con susceptibilidad a la presión, duplicación y mutación puntual PMP-22
- Neuralgia amiotrófica hereditaria, ligada al 17q24-25.
- Tipo II, autosómico dominante locus 12q24.
- Tipo V, autosómico dominante, ligada al cr. 7p.
Tabla 2. Relación entre fenotipo CMT, D-S, NHC y NFSP y genotipo.
| CMT | D-S | NHC | NFSP | |
| Dupl PMP-22 | + | |||
| Mut punt PMP-22 | + | + | + | + |
| Delec PMP-22 | + | |||
| Mut punt P0 | + | + | + | |
| Mut Cx 32 | + | |||
| Mut EGR2 | + | + |
CMT: fenotipo Charcot-Marie-Tooth, D-S: fenotipo Dejerine-Sottas, NHC: neuropatía hipomielinizante congénita, NFSP: neuropatía familiar con susceptibilidad a la presión.

Fig. 1. Esquema representativo de la duplicación de 1.500 kb del locus CMT1A. En la parte superior se indica la región en un cromosoma normal no duplicado . Se observa la localización del gen PMP22 y de los tres loci marcadores utilizados para la detección de la duplicación. En la parte inferior se muestra la región duplicada en tándem en un cromosoma portador de la mutación. CEN y TEL se refieren al centrómero y telómero del brazo corto del cromosoma 17.
Fig 2. Esquema del mecanismo de producción de la duplicación CMT1A y de la deleción en la NFSP. El mecanismo consiste en un mal alineamiento de las secuencia repetitivas CMT1A-REP (REP en la figura) que flanquean la región de 1.500 kb. Ello provoca un entrecruzamiento desigual en la meiosis generándose dos productos en espejo, una duplicaión en tandem y una deleción. El gen PMP22 está indicado por un segmento en verde. En la parte inferior se indican marcadores localizados en la región involucrada y marcadores flanqueantes que no participan en la duplicación o deleción.
Bibliografía
- Charcot JM, Marie P. Sur une forme particulière d'atrophie musculaire progressive, souvent familiale, debutant par les pieds et les jambes atteignant plus tard les mains. Revue de Médicine, Paris 1886; 6:97-138
- Tooth HH. The peroneal type of progressive muscular atrophy. London, H.K. Lewis & Co., Ltd., 1886.
- Dejerine J, Sottas J. Sur la nevrité interstielle, hypertrophique et progressive de l'enfance. Comptes Rendus de la Société de Biologie 1893; 45:63-96.
- Roussy G, Lévy G. Sept cas d´une maladie familiale particulière. Rev Neurol (Paris) 1926; 1:427-450.
- Lapresle J, Salisachs P. Onion bulbs in a nerve biopsy specimen from an original case of Roussy-Lévy disease. Arch Neurol 1973; 29: 346-348.
- Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol 1968; 18: 603-618.
- Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch Neurol 1968; 18: 619-625.
- Thomas PK, Calne DB, Stewart G. Hereditary motor and sensory polyneuropathy (peroneal muscular atrophy). Ann Hum Genet 1974; 38:111-153.
- Buchthal F, Behse F. Peroneal muscular atrophy (PMA) and related disorders. I. Clinical manifestationsas related to biopsy findings, nerve conduction and electromyography. Brain 1977; 100:41-66.
- Behse F, Buchthal F. Peroneal muscular atrophy (PMA) and related disorders. II. Histological findings in sural nerves. Brain 1977; 100:67-85.
- Brust JCM, Lovelace RE, Devi S. Clinical and electrodiagnostic features of Charcot-Marie-Tooth syndrome. Acta Neurol Scand 1978; 58 Supply 68:1-142.
- Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980; 103:259-280.
- Ben Hamida M, Letaief F, Ben Hamida C, Samoud S. Les atrophies péronierès en Tunisie. Étude de 70 Observations pures ou associées à d'autres affections hérédodégénératives. J Neurol Sci 1981; 50:335-356.
- Bouché P, Gherardi R, Cathala HP, Lhermitte F, Castaigne P. Peroneal muscular atrophy. Part 1. Clinical and electrophysiological study. J Neurol Sci 1983; 61:389-399.1974; 38:111-153.
- Humberstone PM. Nerve conduction studies in Charcot-Marie-Tooth disease. Acta Neurol Scand 1972; 48:176-190.
- Salisachs P. Wide spectrum of motor conduction velocity in Charcot-Marie-Tooth disease. An anatomophysiological interpretation. J Neurol Sci 1974;23:25-31.
- Davis CJF, Bradley WG, Madrid R. The peroneal muscular atrophy syndrome (clinical, genetic, electrophysiological and nerve biopsy studies). J Genet Hum 1978; 26:311-349.
- Lyon G. Ultraestructural study of a nerve biopsy from a case of early infantile chronic neuropathy. Acta neuropath. (Berlin) 1969; 13: 131-142.
- Anderson R, X Dennet, Hopkins IJ et al. Hypertrophic interstitial polyneuropathy in infancy. J Pediatrics 1973; 82:619-624.
- Kasman M, Bernstein L, Schulman S. Chronic polyradiculoneuropathy of infancy. Neurology 1976; 26:565-573.
- Kennedy WR, Sung JH, Berry J. A case oh congenital hypomyelination neuropathy. Clinical, morphological, and chemical studies. Arch Neurol1977; 34:337-345.
- Palix C, Coignet J. Un cas de polyneuropathy périphérique neo-natale par amyélinisation. Pediatrie 1978; 201-207.
- Moss R, Sriram S, Kelts A et al. Chronic neuropathy presenting as a floppy infant with respiratory distress. Pediatrics 1979; 64:459-464.
- Guzzetta F, Ferrière G, Lyon G. Congenital hypomyelination polyneuropathy. Pathological findings compared with polyneuropathies starting later in life. Brain 1982; 105: 359-416.
- Hakamada S, Kumagai T, Hara S, et al. Congenital hypomyelination neuropathy in a newborn. Neuropediatrics 1983; 14:182-183.
- Harati Y, Butler IJ. Congenital hypomyelinating neuropathy. J Neurol Neurosurg Psychiatr 1985; 48:1269-1276.
- Johnson MD, Glick AD, Whetsell WO. Central hypomyelination associated with congenital hypomyelinating polyneuropathy: report of an autopsied case. Clin Neuropathology 1989; 8:28-34.
- Bird TD, Ott J, Giblett ER. Evidence for linkage of Charcot-Marie-Tooth neuropathy to the Duffy locus on chromosoma 1. Am J Human Genet 1982; 34:388-394.
- Guiloff RF, Thomas PK, Contreras M, et al. Linkage of autosomal dominant type I hereditary motor and sensory neuropathy to the Duffy locus on chromosome 1. J Neurol Neurosurg Psychiatr 1982; 45:669-674.
- DycK PJ, Ott J, Breanndan Moore S, et al. Linkage evidence for heterogeneity among kinships with hereditary motor and sensory neuropathy type I. Mayo Clin Proc 1983; 58: 430-435.
- Dyck PJ, Chance P, Lebo R, Carney JA.. Hereditary motor and sensory neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Podouslo JF, eds.Peripheral neuropathy. 3rd de. Philadelphia WB. Saunders Company, 1993:1094-1136.
- Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Baas F, Barker DF et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). Neuromuscular disorders 1991; 1:93-97.
- Lupski JR, Montes de Oca-Luna R, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1a. Cell 1991; 66:219-232.
- Raymaekers P, Timmerman V, Nelis E et al. Estimation of the size of the 17p11.2 duplication in Charcot-Marie-Tooth neuropathy type 1 a (CMT 1 A). J Med Genet 1992; 29:5-11.
- Pentao L, Wise CA, Chinault AC, Patel PI, Lupski JR. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeats sequences flanking the 1.5Mb monomer unit. Nature Genetics 1992; 2:292-300.
- Chance PF, Abbas N, Lensch MW, Pentao L, Roa BB, Patel PI, Lupski JR. Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Molec Genet 1994; 3:223-228.
- Mac Millan JC, Upadhyaya M, Harper PS. Charcot-Marie-Tooth disease type 1a (CMT 1a): evidence for trisomy of the region p11.2 of chromosome 17 in South Wales families. J Med Genet 1992; 29:12-13.
- Bellone E, Mandich P, Mancardi GL, Schenone A, Uccelli A, Abbruzzese M, et al. Charcot-Marie-Tooth (CMT) 1a duplication at 17p11.2 in Italian families. J Med Genet 1992; 29:492-493.
- Brice A, Ravisé N, Stevanin G, Gugenheim M, Bouche P, Penet C et al.Duplication within chromosome 17p11.2 in 12 families of French ancestry with Charcot-Marie-Tooth disease type 1a. J Med Genet 1992; 29:807-812.
- Hallam PJ, Harding AE, Berciano J, Barker DF, Malcolm S. Duplication of part of chromosome 17 is commonly associated with hereditary motor and sensory neuropathy type I (Charcot-Marie-Tooth disease type 1). Ann Neurol 1992; 31:570-572.
- Valentijn LJ, Baas F, Wolterman RA, Hoogendijk JE, Van den Bosch Zorn I et al. Identical point mutations of PMP-22 in Trembler-J mouse and Charcot-Marie-Tooth disease type 1a. Nature Genetics 1992; 2:288-291.
- Roa BB, Garcia CA, Suter U, et al. Charcot-Marie-Tooth disease type 1a. Association with a spontaneous point mutation in the PMP-22 gene. N Engl J Med 1993; 329:96-101.
- Roa B, García CA, Pentao L, et al. Evidence for a recessive PMP-22 point mutation in Charcot-Marie-Tooth disease type 1 A. Nature Genetics 1993; 5:189-194.
- Nelis E, Timmerman V, De Jonghe P, et al. Identification of a 5´ splice site mutation in the PMP-22 gene in autosomal dominant Charcot-Marie-Tooth disease type 1.
- Navon R, Seifried B, Shoham Gal-On N, et al. A new point mutation affecting the fourth transmembrane domain of PMP-22 results in severe, de novo Charcot-Marie-Tooth disease. Hum Genet 1996; 97:685-687.
- Marrosu MG, Vaccargiu S, Marrosu G, et al. A novel point mutation in the peripheral myelin protein 22. Neurology 1997; 48: 489-493.
- Suter U, Welcher AA, Özcelik T, Snipes GJ, Kosaras B, Francke U et al. Trembler mouse carries a point mutation in a myelin gene. Nature 1992; 356:241-244.
- Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL et al. A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc Natl Acad Sci U.S.A. 1992; 89:4328-4386.
- Patel PI, Roa BB, Welcher AA, Schoener-Scoth R, Trask BJ, Pentao L et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1a. Nature Genetics 1992; 1:159-165.
- Valentijn LJ, Bolhuis PA, Zorn I, Hoogendijk JE, Van der Bosch N, Hensels GW et al. The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1a. Nature Genetics 1992; 1:166-170.
- Matsunami N, Smith B, Ballard L, Lensch W, Robertson M, Albertsen H et al. Peripheral myelin protein-22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot-Marie-Tooth 1a. Nature Genetics 1992; 1:176-179.
- Timmerman V, Nelis E Van Hul W, et al. The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth duplication. Nat Genet 1992; 1:171-175.
- Palau F, Löfgren A, De Jonghe P, Bort S, Nelis E, Sevilla T et al. Origin of the novo duplication in Charcot-Marie-Tooth disease type 1a: unequal nonsister chromatid exchange during spermatogenesis. Hum Molec Genet 1993; 2:2031-2035.
- Roa BB, DycK PJ, Marks HG, Chance PF, Lupski JR . Dejerine-Sottas syndrome associated with point mutation in the peripheral myelin protein 22 (PMP22) gene. Nature Genetics 1993; 5:269-263.
- Ionasescu VV, Ionasescu R and Searby C. Screening of dominantly inherited Charcot-Marie-Tooth neuropathies. Muscle and Nerve 1993; 16:1232-1238.
- Bort S, Sevilla T, García Planells J et al. Déjerine-Sottas neuropathy associated with de-novo S79P mutation of the peripheral myelin protein 22 (PMP22) gene. Human mutation 1998; 6:S95-S98.
- Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 1993; 72:143-151.
- Nicholson GA, Valentijn LJ, Cherryson AK, Kennerson ML, Bragg TL, De Kroon RM et al. A frame shift mutation in the PMP22 gene in hereditary neuropathy with liability to pressure palsies. Nature Genetics 1994; 6:263-266.
- Hoogendijk JK, Hensels GW, Gabreëls-Festen AAWM, Gabreëls FJM, Janssen EAM, de Jonghe P et al. De-novo mutation in hereditary motor and sensory neuropathy type I. Lancet 1992; 339:1081-1082.
- Palau F, Löfgren A, De Jonghe P, et al. Origin of the novo duplication in Charcot-Marie-Tooth disease type 1a: unequal nonsister chromatid exchange during spermatogenesis. Hum Molec Genet 1993; 2:2031-2035.
- Blair IP, Nash J, Gordon MJ et al. Prevalence and origin of the novo duplications in Charcot-Marie-Tooth disease type 1 A : First report of a de novo duplication with a maternal origin. Am J Hum Genet 1996; 58:472-476.
- Hayasaka K, Himoro H, Wang Y, Takata M, Minoshima S, Shimizu N et al. Structure and chromosomal localization of the gene encoding the human myelin protein zero (MPZ). Genomics 1993; 17:755-758.
- Lemke G and Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell 1985; 40: 501-508.
- Kulkens T, Bolhius PA, Wolterman RA, Kemp S; Nijenhuis S, Valentijn LJ et al. Deletion of the serine 34 codon from the major peripheral myelin protein P0 gene in Charcot-Marie-Tooth disease type 1B. Nature Genetics 1993; 5:35-39.
- Hayasaka K, Himoro M, Sato W, Takada G, Uyemura K, Shimizu N et al. Charcot-Marie-Tooth neuropathy type 1B associated with mutations of the myeline P0 gene. Nature Genetics 1993; 5:31-34.
- Hayasaka H, Himoro M, Sawaishi, Nanao K, Takahashi T, Takada G et al. De novo mutation of the myeline P0 gene in Dejerine-Sottas disease (hereditary motor and sensory neuropathy type III). Nature Genetics 1993; 5: 266-268.
- Hayasaka K, Ohnishi A, Takada G, Fukushima Y, Murai Y. Mutation of the myelin P0 gene in Charcot-Marie-Tooth neuropathy type 1. Biochem Biophys Res Comm 1993, 194:1317-1322.
- Hayasaka K, Takada G and Ionasescu VV. Mutation of the myeline P0 gene in Charcot-Marie-Tooth neuropathy type 1B. Hum Mol Genet 1993: 1369-1372.
- Himoro M, Yoshikawa H, Matsui, MitsuiY, TakahashiM, Kaido M et al. New mutations of the myelin P0 gene in a pedigree of Charcot-Marie-Tooth neuropathy type 1. Biochem Mol Biol Int 1993; 31:169-173.
- Martini R, Zielasek J, Toyka KV, et al. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristics of inherited human neuropathies. Nature Genet 1995; 11: 281-286.
- Chance PF, Matsunami N, Lensch W, Smith B, Bird TD. Analysis of de DNA duplication 17p11.2 in Charcot-Marie-Tooth neuropathy type 1 pedigrees: additional evidence for a third autosomal CMT1 locus. Neurology 1992; 42:2037-2041.
- Ben Othmane K, Hentati F, Lennon F et al. Linkage of a locus (CMT4A) for autosomal recessive Charcot-Marie-Tooth disease to chromosome 8q. Hum Mol Genet 1993; 2:1625-1628.
- Bolino A, Brancolini V, Bono F et al. Localization of a gene responsible for autosomal recessive demyelinating neuropathy with focally folded myelin sheats to chromosome 11q23 by homozygosity mapping and haplotype sharing. Hum Mol Genet 1996; 5: 1051-1054.
- Gambardella A, Bolino A, Muglia M, et al. Genetic heterogeneity in autosomal recessive hereditary motor and sensory neuropathy with focally folded myelin sheats (CMT4B). Neurology 1998; 50: 799-801.
- LeGuern E, Guilbot A, Kessall M et al. Homozygosity mapping of an autosomal recessive form of demyelinating Charcot-Marie-Tooth disease to chromosome 5q23-q33. Hum Mol Genet 1996; 5:1685-1688.
- Kalaydjieva L, Hallmayer J, Chandler D, et al. Gene mapping in Gypsies identifies a novel demyelinating neuropathy on chromosome 8q24. Nat Genet 14:214-217.
- Kalaydjieva L, Nikolova A, Turnev I, et al. Hereditary motor and sensory neuropathy-Lom, a novel demyelinating neuropathy associated with deafness in gypsies. Clinical, electrophisyological and nerve biopsy findings. Brain 1998; 121:399-408.
- Allan W. Relation of hereditary pattern to clinical severity as illustrated by peroneal atrophy. Arch Int Med 1939; 69:1123-1131.
- Gal A, Mucke J, Theile H, Wieacker PF, Ropers HH, Wienker TF. X-linked dominant Charcot-Marie-Tooth disease: suggestion of linkage with a cloned DNA sequence from the proximal Xq. Hum Genet 1985; 70:38-42.
- Goonewardena P, Welihinda J, Anvret M, et al. A linkage study of the locus for X-linked Charcot-Marie-Tooth disease. Clin Genet; 33: 435-440.
- Hahn AF, Brown WF, Koopman WJ, et al. X-linked dominant hereditary motor and sensory neuropathy. Brain 1990; 113:1511-1525.
- Phillips II LH, Kelly TE, Schnatterly P, et al. Hereditary motor-sensory neuropathy (HMSN) : possible X-linked dominant inheritance. Neurology 1985; 35:498-502.
- Rozear MP, Pericak-Vance, Fischbeck K, et al. Hereditary motor and sensory neuropathy, X-linked: a half century follow-up. Neurology 1987; 37:1460-1465.
- Sander S, Nicholson GA, Ouvrier RA , et al.
- Charcot-Marie-Tooth disease: histopathological features of the peripheral myelin protein (PMP22) duplication (CMT1 A) and conexin-32 mutations (CMTX1). Muscle Nerve 1998; 21: 217-225.
- Bergoffen J, Trofatter J, Pericak-Vance MA, et al. Linkage localization of X-linked Charcot-Marie-Tooth disease. Am J Hum Genet 1993; 52:312-318.
- Bergoffen J, Scherer SS, Wang S, Oronzi Scott M, Bone LJ, Paul DL et al. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 1993; 262:2039-2042.
- Fairweather N, Bell C, Cochrane S, Chelly J, Wang S, Mostacciuolo ML et al. Mutations in the connexin 32-gene in X-linked dominant Charcot-Marie-Tooth disease (CMTX1). Hum Mol Genet 1994; 3:29-34.
- Ionasescu V, Searby C, Ionasescu R. Point mutations of the connexin 32 (GJB1) gene in X-linked dominant Charcot-Marie-Tooth neuropathy. Hum Mol Genet 1994; 3:335-358.
- Orth U, Fairweather N, Exler MC, et al. X-linked dominant Charcot-Marie-Tooth neuropathy: valine-38-metionine substitution of connexin 32. Hum Mol Genet 1994; 3: 1699-1700.
- Bone LJ, Dahl N, Lensch MW, et al. New connexin32 mutations associated with X-linked Charcot-Marie-Tooth disease. Neurology 1995; 45:1863-1866.
- Warner LE, Mancias P, Butler IJ, et al. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet 1998; 18:382-384.
- Warner LE, Hilz MJ, Appel SH, et al. Clinical phenotypes of different MPZ mutations may include Charcot-Marie-Tooth 1B, Dejerine-Sottas and congenital hypomyelination. Neuron 1996; 17:451-460.
- Verhagen WIM, Gabreëls-Festen AAWM, van Wensen PJM, et al. Hereditary neuropathy with liability to pressure palsies: a clinical, electroneurophysiological and morphological study. J Neurol Sci 1993; 116: 176-184.
- Jong JGY de. Over families met hereditaire dispositie tot het optreden van neuritiden, gecorreleerd met migraine. Psychiat Neurol Bl 1947; 50: 60-76.
- Behse F, Buchthal F, Carlsen F et al. Hereditary neuropathy with liability to pressure palsies: electrophysiological and histopathological aspects. Brain 1972; 95: 777-794.
- Cruz Martinez A, Perez Conde C, Ramon y Cajal S. A recurrent familial polyneuropathy with the liability to pressure palsies. Specific regards to electrophisiological aspects of 25 members of 7 families. Electromyogr Clin Neurophysiol 1977; 17: 101-124.
- Roos D, Thygesen P. Familial recurrent polyneuropathy. A family survey. Brain 1972; 95: 235-248.
- Lhermitte F, Gautier J, Rosa A. Neuropathie récurrente familiale. Rev Neurol (Paris) 1973; 128:419-424.
- Pou-Serradell A, De Paiva VJ, Almela F, et al. Paralysie recidivante familiale du plexus braquial. Neuropathie tomaculaire. Rev Neurol (Paris) 1992; 148:123-128.
- Verhalle D, Löfgren A, Nelis E, et al. Deletion in the CMT1A locus on chromosome 17p11.2 in hereditary neuropathy with liability to pressure palsies. Ann Neurol 1994; 35: 704-708.
- Reisecker F, Leblhuber F, Lexner R, et al. A sporadic form of hereditary neuropathy with liability to pressure palsies: clinical, electrodiagnostic, and molecular genetics findings. Neurology 1994; 44: 753-755.
- Tyson J, Malcolm S, Thomas PK, et al. Deletions of chromosome 17p11.2 in multifocal neuropathies. Ann Neurol 1996; 39: 180-186.
- Gouider R, LeGern E, Gugenheim M, et al. Clinical, electrophysiologic, and molecular correlations in 13 families with hereditary neuropathy with liability to pressure palsies and a chromosome 17p11.2 deletion. Neurology 1995; 45:2018-2023.
- Timmerman V, Löfgren A, LeGuern E, et al. Molecular genetic analysis of the 17p11.2 region in patients with hereditary neuropathy with liability to pressure palsies (HNPP). Hum Genet 97: 26-34.
- Bort S, Nelis E, Timmerman V, et al. Mutation analysis of the MPZ, PMP22, and Cx32 genes in Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsies patients with Spanish ancestry. Hum Genet 1997; 99:746-754.
- Young P, Wiebusch H, Stögbauer F, et al. A novel frameshift mutation in PMP22 accounts for hereditary neuropathy with liability to pressure palsies. Neurology 1997; 48: 450-452.
- Mariman ECM, Gabreels-Festen AAWM, van Beersum SEC, et al. Prevalence of the 1.5-Mb 17p deletion in families with hereditary neuropathy with liability to pressure palsies. Ann Neurol 1994; 36: 650-655.
- Schenone A, Nobbio L, Mandich P, et al. Underexpression of messenger RNA for peripheral myelin protein 22 in hereditary neuropathy with liability to pressure palsies. Neurology 1997; 48: 445-449.
- Schenone A, Nobbio L, Caponnetto C, et al. Correlation between PMP-22 messenger RNA expresion and phenotype in hereditary neuropathy with liability to pressure palsies. Ann Neurol 1997; 42: 866-872.
- Windebank AJ. Inherited recurrent focal neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Podouslo JF, eds. Peripheral neuropathy. 3er ed. Philadelphia: WB Saunders, 1993: 1137-1148.
- Pellegrino JE Rebbeck TR, Brown MJ, et al. Mapping of hereditary neuralgic amyotrophy (familial braquial plexus neuropathy) to distal chromosome 17q. Neurology 1996; 46: 1128-1132.
- Meadows JC, Marsden CD. A distal form of chronic spinal muscular atrophy. Neurology 1969; 19:53-58.
- Young ID, Harper PS. Hereditary distal spinal muscular atrophy with vocal cord paralysis. J Neurol Neurosurg Psychiatry 1980; 43: 413-418.
- Boltshauser E, Hof E, Spillman T, et al. Dominante spinale muskelatrophie mit stimmbandlahmung und innenohrschwerhorigkeit. Schweiz Arch Neurol Psychiatr 1985; 136: 75-87.
- Pridmore C, Baraitser M, Brett EM, et al. Distal spinal muscular atrophy with vocal cord paralysis. J Med Genet 1992; 29: 197-199.
- Serratrice G, Pellisier JF, Gastaut JL, et al. Amyotrophie spinale chronique avec paralysie des cordes vocales: syndrome de Young et Harper. Rev Neurol (paris) 1984; 140: 657-658.
- Harding AE, Thomas PK. Hereditary distal spinal atrophy. A report on 34 cases and review of the literature. J Neurol Sci 1980; 45: 337-348.
- Sevilla T. Atrofias musculares espinales distales. Neurología 1996 supl; 11: 58-65.
- Harding AE. Inherited neuronal atrophy and degeneration predominantly of lower motor neurons. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Podouslo JF, eds. Peripheral neuropathy. 3er ed. Philadelphia: WB Saunders Company, 1993: 1051-1064.
- Emery AEH. Review: The nosology of the spinal muscular atrophies. J Med Genet 1971; 8:481-495.
- Timmerman V, De Jonghe P, Simokovic S, et al. Distal hereditary motor neuropathy type II (distal HMN II ): mapping of a locus to chromosome 12q24. Hum Mol Gen 5:1065-1069.
- Christodoulou K, Kyriakides T, Hristova AH, et al. Mapping of a distal form of spinal muscular atrophy with upper limb predominance to chromosome 7p. Hum Mol Genet 1995; 4: 1629-1632